Since there is no solvent given in the procedure, you will have to figure one out for yourself. The lab support will provide several solvents for the task: hexane, toluene, dichloromethane, methanol (most likely). You will have to place four spots on your TLC plate: benzoin, the reference compound, the crude and the purified product. Ideally, the Rf-values should vary by 0.3-0.4. This might be accomplished using one solvent or a solvent mixture. If you use a single solvent (X) first, make sure to try at least two different solvent mixture as well i.e. one with a more non-polar solvent (Y) added (X:Y=5:1) and one with a little polar solvent (Z) added (X:Z=10:1).
Part II: NMR simulations (dry lab!)
The assignment is done during the second half of the lab period in SLC.
Using ACD labs Software, simulate and analyze some of the the spectra for the following compounds.
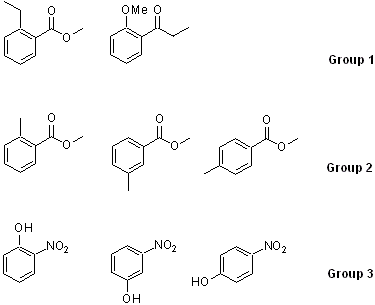
Generating the molecules
In order to draw these compounds, you will have to use ChemSketch (the drawing module of the software). Try to make use of the existing templates available in the program e.g. benzene, cyclopentane, etc. Otherwise, the drawing process is similar to the one in Spartan. However, you cannot draw the molecule in Spartan and then copy it over!
To expand an area, push both mouse buttons. A crosshair appears on the screen and a second screen on the top. Define the left edge by moving the cursor to the left side and clicking the left mouse button. Then hold the left mouse button and drag the cursor to the right and click again. A dark gray field will move with the cursor to highlight the area to be expanded. Alternately, you can also drag the markers on the top window to define the range.
1H-NMR spectra:
1. After you draw the molecules, generate the proton spectrum (ACD/labs 1H-NMR Spectrum Generator) of the compound (the default frequency should be 400 MHz at this point, it can be changed in the Options Menu in the H-NMR module).
a. For group 1, analyze the peak locations and the splitting patterns for the aliphatic range.
b. For group 2, analyze the splitting pattern of the aromatic range and rationalize the location for the Me-signal on the ring. Compare the obtained spectra with the literature spectra that can be found on www.sigmaaldrich.com.
c. For group 3, analyze the splitting pattern of the aromatic range and rationalize the location for the OH signal.
2. Next, change the Default Basic Frequency to 60 MHz and recalculate the spectra for group 2. What changes? How can you explain the change?
3. Using the molecules of group 2, activate the Integration feature in the Tools-menu. Can you account for its shape and the steps observed?
13C-NMR spectra:
1. You can similate the 13C-NMR spectrum in the fashion (ACD/labs CNMR Spectrum Generator). Compare the different substitution patterns in the 13C-NMR spectrum of the molecules in group 2. (All generated spectra are proton-decoupled.)
2. If you check the "Off-Resonance" feature (in the Tools-Menu), the decoupling feature is deactivated. How do you the spectra change? Which conclusion can you make from this type of spectra?
3. The program also allows you to do a "virtual lab". Prepare a solution of 10 mg of o-nitrophenol in 1 mL of CDCl3. How does the spectrum look like? Then change the concentration to 100 mg/mL CDCl3 and 1 mL CDCl3. Which changes do you observe in the spectrum?
Thinking questions
1. When do you think such a program is useful to predict chemicals shift accurately? When do you expect it to fail?
2. The version provided in the SLC is only able to deal with 50 atoms. Assuming you have a relatively large molecule, which is very symmetric, how would you use the program to get information?