ELECTRON TRANSFER:
Christopher Bardeen*
Sometimes You Can Go Home AgainFrom simple nucleophilic substitution reactions in organic chemistry to photosynthesis, electron transfer is a basic element of chemical reactions in liquids (1). The theoretical framework for understanding electron transfer rates in systems near equilibrium was developed by Marcus and verified experimentally by many workers. The advent of ultrafast lasers has provided physical chemists with a tool for studying how these systems evolve under nonequilibrium conditions. Such studies have revealed molecular details of how electrons move in dense media. On page 462 of this issue, Martini et al. go one step further, providing evidence that femtosecond pulses may be used not only to observe electron transfer dynamics but to control them as well (2).
The system we are concerned with here is an electron embedded in a molecular liquid. An electron is arguably the simplest possible reactant in a condensed phase environment because it lacks the intramolecular vibrational modes of a molecular solute. A dissolved electron can be prepared by a variety of methods in a variety of solvents, but its essential characteristics remain the same.
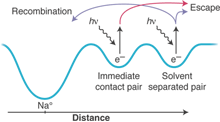
Wayward electrons. In the sodide/tetrahydrofuran system, the initially excited electron can exist in an immediate contact pair or a solvent-separated pair. Electrons in immediate contact pairs will leave when the control pulse is applied, but some solvent-separated pairs recombine with the sodium atom.
At equilibrium, the electron is nestled in a solvent cavity, kept in place by the solvent dipoles. Absorption of a photon excites the electron into a delocalized state, whose wave function may then relocalize to different sites in the solvent that are spatially separated from the original low-energy site. As time goes on, many electrons will lose energy and find their way back to their original cavities, a process known as recombination. But some will escape into the solvent, never to return.
Both types of events are examples of electron transfer from one solvent site to another, and both can be followed by observing the transient absorption spectra of the electron because its wave function, which depends on its spatial extent and environment, also determines its spectral behavior. By using femtosecond pulses, one can observe the creation, relaxation, and recombination of the wayward electron in detail.
The apparent simplicity of the solvated electron has made it the subject of many studies. Pioneering work by Eisenthal and Antonetti established that electrons created by direct ionization of simple solutes in water undergo a complex relaxation process that involves distinct solvation states [reviewed in (3, 4)]. More recent studies (5-10) have concentrated on the dynamics of electron recombination in water under a variety of conditions. In particular, Barbara and co-workers at the University of Texas have clarified the nature of the electron's excited states by quantifying their spatial extent and relaxation times in water (9). Very recently, they have demonstrated how recombination can be suppressed by direct excitation of the solvated electron to very delocalized conduction band states (10).
In Martini et al.'s experiment, the electron is originally localized in a cavity that it shares with a sodium atom, forming a "sodide" ion in room temperature tetrahydrofuran. As in water, the escape probability of the electron can be enhanced by providing extra energy from a second pulse. But the authors do not stop there. They show that by delaying the second pulse an appropriate amount of time, one can enhance the rate of recombination as well.
To accomplish this, the authors exploit a subtlety of the sodide/tetrahydrofuran system, namely that the initially excited electron can exist in two different sites: a closely held immediate contact pair and a more distant solvent-separated pair (see the figure). The two species have different relaxation times, and the key is to wait long enough so that the control pulse excites only the more separated sites. Electrons that originally stayed close to home in immediate contact pairs will leave when the control pulse is applied, but some that originally ventured farther afield in solvent-separated pairs can be persuaded to return more quickly. The authors have thus found an experimental handle to control whether an electron continues to wander off into the solvent or returns home to its original solvent cavity.
It is important to point out that the observed enhancement in recombination rate is small and that the net effect of adding energy to the system with the control pulse, no matter what the delay, is to enhance the escape probability. In other words, the control pulse provides a transient acceleration of the recombination rate, but the time-integrated reaction yield only changes in one direction--toward enhanced escape.
In some sense, this is academic because the main goal of the research is the characterization of the electron's dynamics in liquids. The authors point out that this "control" experiment confirms their previous hypothesis about the existence of two states of the electron. But to what extent can electron dynamics in complex fluids be controlled?
Wasielewski and co-workers have demonstrated that femtosecond pulse sequences can be used to turn electron transfer on and off in designed molecular assemblies (11). And recent advances in pulse shaping and feedback control have demonstrated impressive results in the control of gas phase molecular dissociation (12). Can such an approach be extended to find a combination of pulse delays and excitation wavelengths that will create and then turn off a delocalized electron state in a liquid, in effect creating an ultrafast polarizable switch? The results reported here suggest that it may be worth trying.

References
- P. J. Rossky, J. D. Simon, Nature 370, 263 (1994).
- I. B. Martini, E. R. Barthel, B. Schwartz, Science 293, 462 (2001).
- H. Lu, F. H. Long, K. B. Eisenthal, J. Opt. Soc. Am. B 7, 1511 (1990).
- Y. Gaudel et al., J. Opt. Soc. Am. B 7, 1528 (1990).
- J. Peon et al., J. Phys. Chem. A 103, 2460 (1999).
- V. H. Vilchiz et al., J. Phys. Chem. A 105, 1711 (2001).
- A. Baltuska, M. F. Emde, M. S. Pshenichnikov, D. A. Wiersma, J. Phys. Chem. 103, 10065 (1999).
- M. Assel, R. Laenen, A. Laubereau, J. Chem. Phys. 111, 6869 (1999).
- T. W. Kee, D. H. Son, P. Kambhampati, P. F. Barbara, J. Phys. Chem., in press.
- D. H. Son, P. Kambhampati, T. W. Kee, P. F. Barbara, Chem. Phys. Lett., in press.
- A. S. Lucas, S. E. Miller, M. R. Wasielewski, J. Phys. Chem. B 104, 931 (2000).
- R. J. Levis, G. M. Menkir, H. Rabitz, Science 292, 709 (2001).
The author is in the Department of Chemistry, University of Illinois, Urbana, IL 61801, USA. E-mail: bardeen@scs.uiuc.edu